




Vom Gaming zu wegbereitender Biologie: KI und das Proteinfaltungsproblem Understand article
KI-Systeme wurden entwickelt, um Menschen in Spielen zu besiegen. Wie können sie helfen die Geheimnisse von Proteinfunktionen zu entschlüsseln?
Proteine falten sich zu komplexen dreidimensionalen (3D) Strukturen. Diese Strukturen zu bestimmen ist entscheidend, um biologische Prozesse zu verstehen. Dazu sind allerdings zeitaufwändige, teure Experimente nötig. Wissenschaftler versuchen schon seit ungefähr 50 Jahren, Proteinfaltungen rechnerisch mit Computern vorherzusagen, aber der Fortschritt war langsam und begrenzt. Nach der Entwicklung von künstlichen Intelligenzen (KI) für Spiele, die Menschen in dem abstrakten Brettspiel Go oder Blizzard’s Starcraft besiegt haben, hat das zu Google gehörende Unternehmen DeepMind nun kürzlich AlphaFold2 entwickelt: Ein KI-System, das in der Lage ist, viele Proteinstrukturen mit experimenteller Genauigkeit vorherzusagen. Dieser Artikel erklärt, wie wichtig dieser aufregende Durchbruch ist.
Wie sehen Proteine aus?
Proteine sind der Schlüssel zu fast allen biologischen Prozessen. Es gibt ungefähr 20 000 verschiedene Proteine im menschlichen Körper und Millionen auf der Erde. Jedes einzelne hat eine einzigartige Struktur.
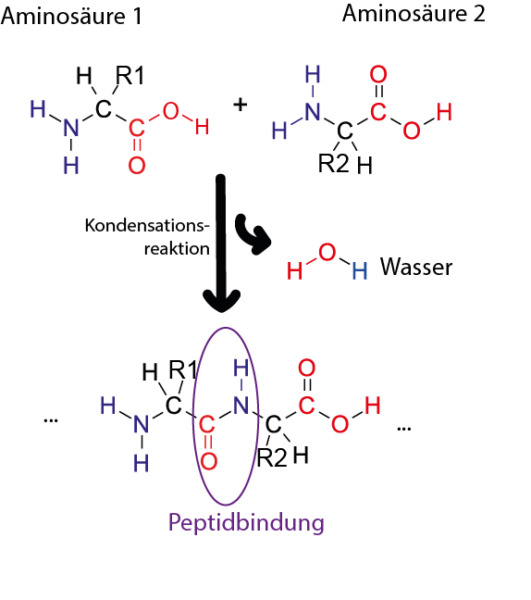
Abbildung von Simone Heber
Die Primärstruktur eines Proteins besteht aus einer Sequenz von Aminosäuren, die durch Peptidbindungen verbunden sind. Proteine sind aus 20 verschiedenen Aminosäuren aufgebaut und deren Sequenz ist in unserer DNS kodiert. Teile einer Aminosäurekette können sich zu Sekundärstrukturen falten, wie α-Helices und β-Faltblättern. Diese Sekundärstrukturen können dann miteinander interagieren, um komplexe 3D-Formen (die Tertiärstruktur) anzunehmen. Aufgrund der Art wie Proteinketten sich falten, können Aminosäuren, die in der Sequenz weit voneinander entfernt sind, in der 3D-Struktur nah bei einander liegen.
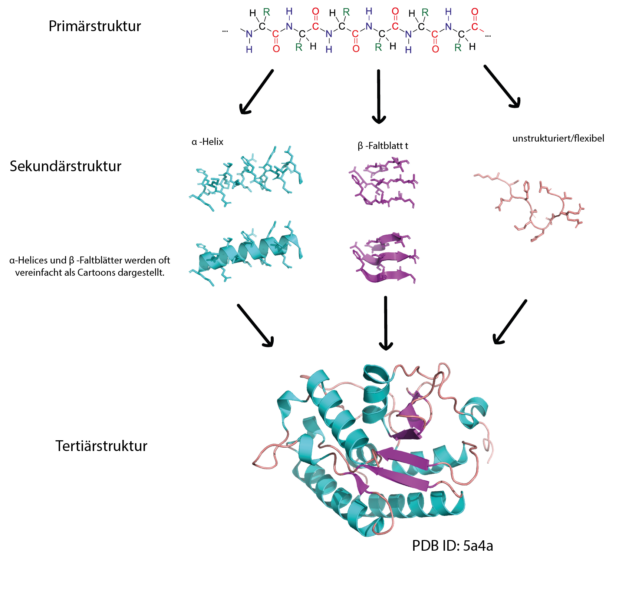
Abbildung von Simone Heber
Die 3D-Struktur eines Proteins bestimmt seine Funktion und falls bei der Faltung etwas schiefläuft, kann das zur Fehlfunktion und Krankheit führen. Falsch gefaltete Proteine spielen zum Beispiel bei Alzheimer- und Parkinson-Erkrankungen eine Rolle.
Die 3D-Strukturen von Proteinen aufzuklären ist grundlegend, um die fundamentalen Funktionen des Lebens zu verstehen und kann dabei helfen, Krankheiten zu bekämpfen. Zum Beispiel kann die Struktur eines Proteins, das mit einer Krankheit in Verbindung steht, den Entwurf eines Wirkstoffs anleiten.
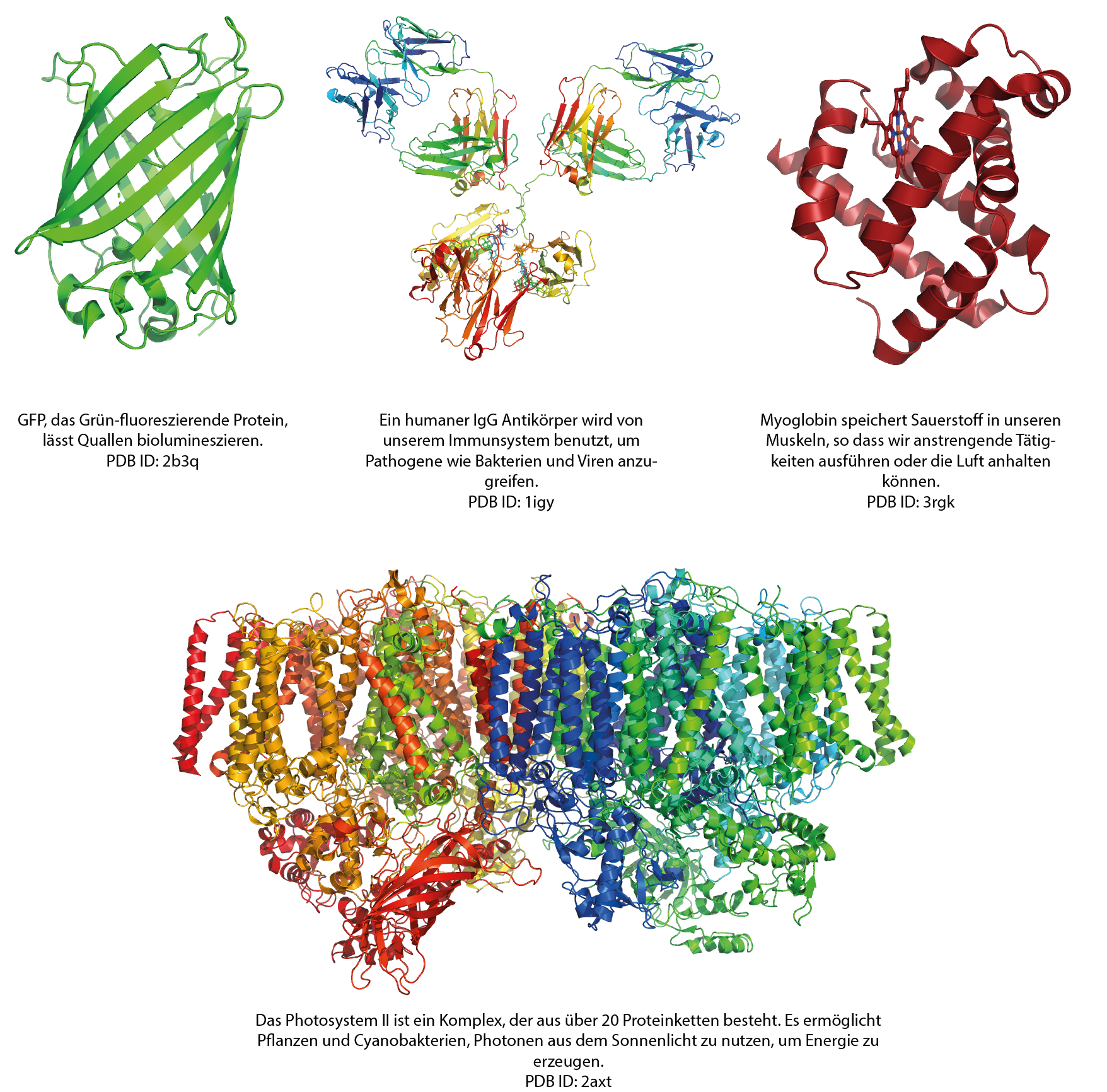
Abbildung von Simone Heber
Experimentelle Methoden zur Proteinstrukturbestimmung
1962 erhielten Max Perutz und John Kendrew (der später einer der Gründer des EMBL und erster Direktor wurde) den Chemie-Nobelpreis für die Bestimmung der ersten Proteinstruktur, der von Myoglobin, durch die Röntgenkristallographie. [1]
Bei der Röntgenkristallographie werden Proteinkristalle mit sehr starken Röntgenstrahlen beschossen, zum Beispiel aus einer Synchrotron-Lichtquelle. Manche Universitäten besitzen auch kleinere, schwächere Instrumente. Der Proteinkristall beugt die Röntgenstrahlen und das Beugungsmuster ermöglicht es Wissenschaftlern, die Proteinstruktur zu berechnen.

Christian Hendrich, GNU Free Documentation License, Version 1.2

Foto von Simone Heber
Foto von Simone Heber
Eine zweite Methode ist die Kernspinresonanzspektroskopie (Nuclear Magnetic Resonance spectroscopy, NMR spectroscopy), welche die Magnetfelder von Atomkernen misst. Diese Magnetfelder werden von der Umgebung der Atome beeinflusst. Bei Atomen in Proteinen enthalten sie also Informationen darüber, welche Atome nah beieinander sind. Für diese Arbeit wurde 2002 der Chemie-Nobelpreis verliehen. [2]

Foto von Simone Heber.
Eine dritte leistungsfähige Methode ist die Kryoelektronenmikroskopie (Kryo-EM), welche man auf schockgefrorene Proben anwendet, wofür 2017 der Chemie-Nobelpreis verliehen wu. [3] In der Elektronenmikroskopie werden Elektronen anstatt Licht benutzt, wodurch noch kleinere Details aufgelöst werden können.
Diese Methoden ermöglichten es Wissenschaftlern, schon mehr als 150 000 Proteinstrukturen zu bestimmen und sie in der Proteindatenbank PDB zu veröffentlichen. [4] Aber diese Methoden sind langsam, teuer, und oft durch die Natur der Proteine selbst eingeschränkt. Wissenschaftler versuchen manchmal jahrelang eine Proteinstruktur zu lösen, ohne Garantie auf Erfolg.
Warum können wir die 3D Struktur eines Proteins nicht basierend auf seiner Primärstruktur ausrechnen?
1972 erhielt Christian B. Anfinsen den Chemie-Nobelpreis, weil er zeigen konnte, dass die Sequenz eines Proteins seine Struktur bestimmt. [5] Die Idee, die 3D-Struktur eines Proteins ausgehend von seiner Aminosäuresequenz zu berechnen, gibt es also schon seit ungefähr 50 Jahren. Und seit die menschliche DNS-Sequenz bekannt ist, sind die Primärstrukturen von Proteinen allgemein verfügbar. Warum war das also bisher so wenig erfolgreich?
Eine Aminosäurekette kann theoretisch eine riesige Menge an Tertiärstrukturen annehmen: Für ein Protein mit 100 Aminosäuren gibt es schätzungsweise 10300 (das ist eine 10 mit 299 Nullen!) mögliche Strukturen. In der Natur falten sich Proteine normalerweise zu ihrer stabilsten Struktur. Diese Struktur mit minimaler Energie kann berechnet werden, aber zum Vergleichen aller möglicher Strukturen benötigt man eine enorme Rechenleistung. Zu den Bemühungen zur Lösung des „Faltungsproblems“ gehört unter anderem das verteilte System Folding@home [6], ein Projekt, welches mittlerweile eines der schnellsten Rechensysteme der Welt ist, indem es sich Rechenkapazität von Freiwilligen leiht. Jeder kann mitmachen und Wissenschaftlern helfen, indem er ungenutzte Rechenleistung seines Computers, Smartphones oder seiner PlayStation3 beisteuert!
Der CASP. Wettbewerb und sein Gewinner von 2020, AlphaFold
Das Forum „Critical Assessment of Protein Structure Prediction” (CASP) [7] wurde 1994 gebildet und veranstaltet alle zwei Jahre einen Wettbewerb, in dem Wissenschaftler Computerprogramme nutzen, um Proteinstrukturen vorherzusagen, die zwar schon experimentell bestimmt, aber noch nicht veröffentlicht wurden. Keine davon war jedoch bisher in der Lage, genaue Proteinstrukturvorhersagen zu treffen.
2018 trat DeepMind [8, 9 ] dem Wettbewerb bei und gewann ihn mit der künstlichen Intelligenz (KI) AlphaFold. Im Wettbewerb von 2020 machte AlphaFold2 nochmal einen riesigen Sprung nach vorn. Es sagte über 90 % der Proteinstrukturen mit experimenteller Genauigkeit vorher und ließ damit seine Mitstreiter auf der Strecke. [9–11 ]
Das Unternehmen DeepMind gehört zu Google und ist am besten bekannt für seine KIs, die menschliche Spieler in Spielen wie Schach, Go und StarCraft besiegen können. 2017 schlug seine KI AlphaGo den weltbesten Go-Spieler. Die KI wurde dann umfunktioniert und lernte Schach zu spielen ohne jegliches menschliche Zutun.
AlphaFold2 ist eine deep-learning basierte KI. Der KI wurden über 100 000 bekannte Proteinfaltungen zum Training gegeben; damit machte man sich die Arbeit hunderter Wissenschaftler zunutze. Dann nutzt die KI die Muster, die sie aus dem Trainingssatz gelernt hat, um innerhalb weniger Tage genaue Proteinstrukturen vorherzusagen.
Was bedeutet das für Wissenschaft und Gesellschaft?
DeepMind will AlphaFold nutzen, um Strukturen von Proteinen zu bestimmen, die in Krankheiten eine Rolle spielen, und damit helfen, Medikamente zu entwickeln. Strukturinformationen können auch die Entwicklung von Enzymen unterstützen, die Plastik abbauen oder Biotreibstoffe herstellen können. Da die experimentelle Bestimmung von Proteinstrukturen teuer und zeitaufwändig ist, könnten genaue Strukturvorhersagen die Forschung drastisch beschleunigen und ihre Kosten reduzieren.
Abbildung von Simone Heber
Dennoch bleiben Fragen offen, denn obwohl Machine-Learning-Methoden Strukturvorhersagen liefern können, erklären sie nicht, wie Proteine sich falten. Wenn ein Protein all seine 10300 möglichen Strukturen ausprobieren würde, bräuchte es länger als das Alter des Universums um sich zu falten. In der Natur können sich Proteine aber in Millisekunden falten. Dies wird als das „Levinthal Paradoxon“ bezeichnet, nach Cyrus Levinthal, der es 1969 formulierte.
Des Weiteren besitzen Proteinstukturen eine gewisse Flexibilität, beispielsweise wenn sie an ein anderes Protein oder einen Arzneiwirkstoff binden. Dann hängt ihre Faltung nicht mehr allein von der Primärsequenz ab. Das bedeutet, dass selbst bei präzisen Vorhersagen von Einzelstrukturen experimentelle Strukturbestimmungen und funktionelle Studien wichtig bleiben werden.
Präzise Strukturvorhersagen haben aber das Potential, den wissenschaftlichen Fortschritt zu beschleunigen und viele teure, arbeitsaufwändige Experimente einzusparen. Vorhersagen können das Design von Experimenten steuern und das wissenschaftliche Vorgehen optimieren, so dass sich Wissenschaftler schneller fortgeschritteneren Problemen zuwenden können.
References
[1] Chemie-Nobelvorlesung 1962, Speed read: https://www.nobelprize.org/prizes/chemistry/1962/speedread/
[2] Chemie-Nobelvorlesung 2002, press release: https://www.nobelprize.org/prizes/chemistry/2002/press-release/
[3] Chemie-Nobelvorlesung 2017, press release: https://www.nobelprize.org/prizes/chemistry/2017/press-release/
[4] Homepage der Protein Data Bank: http://www.rcsb.org/
[5] Chemie-Nobelvorlesung 1972, press release: https://www.nobelprize.org/prizes/chemistry/1972/press-release/
[6] Homepage des Folding@home Computing Projekts: https://foldingathome.org/
[7] Protein Structure Prediction Center: https://predictioncenter.org/
[8] Alphafold Webpage: https://deepmind.com/research/case-studies/alphafold
[9] Ein Nature News Artikel über Alphafold: https://www.nature.com/articles/d41586-020-03348-4
[10] MIT technology review: https://www.technologyreview.com/2020/11/30/1012712/deepmind-protein-folding-ai-solved-biology-science-drugs-disease/
[11] AlphaFold2 faus dem Blickwinkel eines Forschers: https://www.asbmb.org/asbmb-today/science/120520/ai-makes-huge-progress-predicting-how-proteins-fol
Resources
- Evolution und Biochemie lehren mit online biologischen Datenbanken: Tenorio G (2014) Using biological databases to teach evolution and biochemistry . Science in School 29: 30-34.
- Entdecke, wie die Struktur des grün fluoreszierenden Proteins seine Lichtemissionseigenschaften bestimmt: Furtado S (2009) Painting life green: GFP . Science in School 12:19-23.
- Mehr über Proteinkristallographie und das ESRF: Cornuéjols D (2009) Biological crystals: at the interface between physics, chemistry and biology . Science in School 11: 70-76.
- Mehr über die bioinformatische Datenspeicherung am EMBL-EBI: Stroe O (2018) Bioinformatics: the new ‘cabinet of curiosities’. Science in School 44: 20-24.
- Hier im Video ansehen, wie Wissenschaftler das Rätsel der Proteinfaltung lösen (auf Deutsch)
- Ein Cartoon über Proteinstruktur und -faltung
Author(s)
Dr Simone Heber ist Postdoc am EMBL in Heidelberg, wo sie Interaktionen zwischen Proteinen und RNA während der Entwicklung von Eizellen erforscht. Sie erhielt ihren Doktorgrad in einem strukturbiologischen Labor, in dem sie Röntgenkristallographie und NMR-Spektroskopie nutzte um zu untersuchen, wie ein neuronales Protein bestimme RNAs erkennt und dadurch zu unserer Erinnerungsbildung und zum Lernen beiträgt.

Foto von Simone Heber.
Review
Die Behandlung von Krankheiten wie Alzheimer und Krebs oder Infektionen hängt stark davon ab, neue molekulare Ziele für das Design neuer Medikamente zu identifizieren. Computertechnologien und künstliche Intelligenzen verkürzen die benötigte Zeit und reduzieren die Kosten hierfür. Dieser Artikel vermittelt unseren Schülern das Wesen der interdisziplinären Zusammenarbeit in der Forschung als eine Möglichkeit, Krankheiten effizienter zu bekämpfen.
Der Artikel ist nützlich für die Lehre der Biologie und Chemie, um die Wichtigkeit von molekularen 3D-Strukturen vorzustellen, ganz besonders der Faltung von Aminosäuresequenzen in Proteinen, und zu zeigen, wie künstliche Intelligenz und Bioinformatik uns helfen können, die versteckten Informationen in unseren Zellen und Molekülen zu entschlüsseln.
Jesús López Alonso, Biologie- und Geologielehrer, IES Gil y Carrasco-Ponferrada, Spain