




Proteínas letais: priões Understand article
Traduzido por Artur Melo. Desde a epidemia da ‘doença das vacas loucas’ nas décadas de 80 e 90 do século passado, e da emergência da sua equivalente humana, variante da doença de Creutzfeld-Jacob, tem havido um enorme esforço de investigação sobre os priões, os agentes responsáveis…

/ iStockphoto
Poderá existir uma epidemia oculta, à espera de acontecer, com milhões de pessoas já infectadas; não podemos evitá-la ou curá-la, e nem sequer diagnosticá-la até que os sintomas fatais surjam.
A doença é a variante da doença de Creutzfeld-Jacob (vCJD), uma num grupo de doenças conhecidas como encefalopatias espongiformes transmissíveis (TSEs), as quais são provocadas e transmitidas através de formas anormais de proteínas priónicas. Exemplos de TSEs incluem não apenas a vCJD, mas também ‘scrapie’ (paraplexia enzoótica) em ovinos, a encefalopatia espongiforme bovina (BSE ou doença das vacas loucas) em gado bovino e kuruw1 em humanos. Estas doenças criam espaços de grandes dimensões, preenchidos por líquido, no tecido cerebral porque a acumulação de priões anormais provoca a morte dos neurónios (células nervosas). Estes espaços característicos atribuem às doenças os seus nomes: encefalopatias (doenças cerebrais) espongiformes (com aspecto de esponja).
As TSEs afectam o sistema nervoso central, com sintomas que incluem problemas com a coordenação e o equilíbrio, tremores e movimentos espasmódicos incontroláveis. Nos humanos, as TSEs também provocam alteração de personalidade e depressão, e os doentes podem sentir confusão, problemas de memória e insónia. À medida que as doenças progridem, a maioria das funções mentais desaparecem, incluindo a capacidade de falar. Todas as TSEs são fatais e, por enquanto, não há cura.

priónicas (laranja) num
animal infectado com ‘scrapie’
Imagem cortesia de R.
Dourmashkin / Wellcome Images
Os priões são proteínas específicas que se encontram principalmente no sistema nervoso, onde – nas suas formas normais – podem desempenhar funções importantes. Por exemplo, estudos em lesmas marinhas, Aplysia, sugerem que os priões desempenham um papel crucial na formação da memória (Si et al., 2010). Os priões infecciosos são formas anormais (aberrantes) de proteínas priónicas que se replicam dentro do hospedeiro forçando as proteínas normais do mesmo tipo a adoptar a estrutura aberrante. Segue-se um efeito dominó pelo qual um pequeno número de priões aberrantes podem modificar muitos priões normais e, eventualmente, conduzir à doença. Como os priões aberrantes formam amilóides – agregados de proteínas – nas células, as células morrem, criando buracos no cérebro.
Os priões são o único caso conhecido de proteínas patogénicas (que provocam doenças) que se auto-propagam, e são capazes de provocar doenças graves, apesar de parecerem ser simples moléculas proteicas: ao contrário das bactérias, vírus e os outros agentes patogénicos conhecidos, não têm nenhuma informação codificada em ácidos nucleicos (ADN ou ARN) sobre como invadir e replicar-se dentro do hospedeiro. Existe ainda um véu de mistério em torno dos priões, de como se replicam exactamente e atravessam a barreira hemato-encefálica e a barreira da espécie – ou seja, infectam diferentes espécies de hospedeiros.
Foi na década de 1960 que os investigadores descobriram pela primeira vez que os agentes causadores das TSE pareciam não ter ácidos nucleicos; Tikvah Alper sugeriu que o agente era uma proteína. Esta ideia pareceu herética porque todos os outros agentes causadores de doença continham ácidos nucleicos e a sua virulência e patogénese era determinada geneticamente.

bacteriologista
Robert Koch (1843-
1910)
Imagem cortesia de
Wellcome Library,
London
Contudo, três décadas de investigações subsequentes, desenvolvidas principalmente por Stanley Prusiner, que foi galardoado com o Prémio Nobel da Medicina em 1997 pelo seu trabalho com priões e TSEs, levaram à aceitação geral desta ‘hipótese proteica’’w2.
Porém, ainda há quem acredite que as doenças provocadas por priões são, na realidade, provocadas por vírus não convencionais e que os priões são apenas parte destes misteriosos vírus. Os postulados de Koch indicam o que é necessário para provar que um certo agente provoca uma determinada doença; um dos passos necessários é usar esse agente para induzir a doença num organismo saudável. Para provar que a TSE é de facto provocada por um prião, os priões aberrantes, isolados e purificados, devem ser utilisados para transmitir a doença. Só em Fevereiro de 2010 é que este procedimento foi executado, acrescentando evidências substanciais que apoiam a hipótese proteica (Wang et al., 2010).
Porque temem alguns cientistas uma epidemia de vCJD?
A estirpe de prião mais preocupante é a que provoca a vCJD – uma forma de doença das vacas loucas que atravessou a barreira das espécies para infectar humanos (ver caixa). O primeiro caso de vCJD apareceu no Reino Unido em 1996 onde, desde então, vitimou 168 pessoas, com um pico em 2000 quando morreram 28 pessoasw3, e houve um pequeno número de casos noutros países, incluindo a França, Itália, Irlanda, Canadá e os EUA. Números exactos de pessoas infectadas ou que morrem desta doença a nível mundial não são conhecidos e apesar de não haver a certeza de como as pessoas são infectadas, pensa-se que a principal via de infecção é através da ingestão de carne infectada.
| Reino Unido | 172 (4) |
|---|---|
| França | 25 (0) |
| República da Irlanda | 4 (0) |
| Itália | 2 (1) |
| EUA |
3 (0) |
| Canadá | 1 (0) |
| Arábia Saudita | 1 (1) |
| Japão | 1 (0) |
| Holanda | 3 (0) |
| Portugal | 2 (0) |
| Espanha | 5 (0) |
A maioria das outras doenças causadas por priões, incluindo a CJD ‘normal’, são genéticas ou com motivos desconhecidos, e afectam as pessoas idosas. As variantes da CJD diferem da CJD porque afectam jovens e os priões aberrantes se encontram não apenas no cérebro mas também em tecidos como o sangue, amígdalas e apêndices; este facto permite novos modos de transmissão à população em geral e não apenas aos idosos.
Por exemplo, os priões podiam ser transmitidos através de transfusões sanguíneas. No Reino Unido, o sangue doado é leucodepletado (os leucócitos, que podem abrigar priões infecciosos, são removidos). Muitos outros países (por exemplo, a Alemanha) proíbem doações de sangue de pessoas que tenham vivido no Reino Unido.
A doença pode também ser transmitida através de instrumentos cirúrgicos: os priões infecciosos são resistentes a temperaturas elevadas, irradiação e tratamentos químicos vulgares que destroem outros agentes patogénicos. Estimativas de quão sério é o risco variam.
Além disso, também foram encontrados priões nos tecidos do epidídimo e no plasma seminal de carneiros (Gatti et al., 2002). Tais constatações criaram preocupações relativamente à infecção de priões através da doação de esperma e levaram os EUA a rejeitar a doação de esperma por europeus ou homens que tenham vivido na Europa. Todavia, um estudo de peritos a nível mundial estimou que a probabilidade da transmissão de priões pelo esperma era inferior a 1:10 000 000 (Mortimer & Barratt, 2006).
Mas o contacto com tecidos – quer através de esperma, de sangue ou de instrumentos cirúrgicos – não é a única fonte potencial de transmissão: quase todos nós consumimos leite ou produtos lácteos.
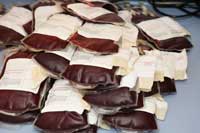
Imagem cortesia de choja /
iStockphoto
Imagem cortesia de UCL /
Wellcome Images
Imagem cortesia de
ImageMediaGroup /
iStockphoto
Em 2006, uma equipa de cientistas da Suíça detectou níveis reduzidos das formas normais de priões em leite adquirido em lojas europeias (Franscini et al., 2006). Outro estudo descobriu que os priões aberrantes conseguiam replicar-se nas glândulas mamárias de ovelhas infectadas com ‘scrapie’ (Ligios et al., 2005). Em conjunto, estes resultados sugerem que os priões aberrantes podem estar presentes no leite de animais que tenham doenças provocadas por priões. Como os sintomas destas doenças demoram vários anos a manifestar-se, os priões aberrantes podem encontrar-se no leite que é vendido, antes de ser diagnosticada a doença nos animais. Contudo, apesar de ainda não ser possível avançar com uma estimativa clara do risco, aceita-se geralmente que o leite é seguro até prova em contrário.

por priões: leite
Imagem cortesia de
DaveAlan / iStockphoto
Parece, por isso, que podemos contrair vCJD através de várias vias, apesar do risco ser provavelmente baixo. Mas estará cada um de nós sujeito ao mesmo risco? Vários estudos mostraram que a vCJD apenas afecta pessoas que possuem certas variantes (alelos) de um determinado gene (prnp); as outras pessoas parecem ser resistentes ou não apresentam os sintomas tão rapidamentew4. Torna-se preocupante cerca de 40% da população de europeus ocidentais e norte americanos e 92% da população japonesa ser homozigótica para estes alelos susceptíveis. Além disso, a investigação sobre o kuru sugere que mesmo os indivíduos sem a susceptibilidade alélica podem ficar infectados, demorando apenas mais tempo a manifestar os sintomas. Por isso, talvez sejamos todos susceptíveis à infecção priónica.
Devido a todas estas possíveis vias de transmissão e às incertezas relativamente à sua importância, os resultados dos estudos que prevêm o número de mortes provocadas por vCJD no Reino Unido, vá de várias centenas a mais de dez milhões de pessoas. Mesmo que agora pareça que a epidemia de vCJD esteja a diminuir, isso pode dever-se apenas a que muitas pessoas infectadas ainda estejam a desenvolver sintomas. Ninguém tem a certeza. Investigação em curso permitirá elucidar qual o risco das doenças priónicas para a saúde pública.
BSE (doença das vacas loucas)
A BSE despertou a atenção dos cientistas em 1986, quando os primeiros casos de uma nova doença neurológica no gado foram descobertos no Reino Unido. A causa foi atribuída ao uso de derivados de carne na alimentação do gado. Na epidemia de BSE que se sucedeu, 181 376 casos de BSE foram registados no RU até Novembro de 2002, e milhões de animais foram abatidos para travar a epidemia. Os primeiros casos fora do RU apareceram em 1989 e, desde então, têm ocorrido vários milhares de casos noutros países, incluindo a maior parte dos países europeus, os EUA, Canadá, Japão e Israel. No entanto, a alimentação de gado com derivados de origem animal foi proibida no RU em 1988 e estão a ser usados procedimentos de fiscalização rigorosa no acompanhamento da BSE.
Os casos de BSE tiveram um pico no RU em 1992 (37 280 casos) e diminuíram drasticamente desde então (12 casos em 2009). O número de casos noutros países tiveram um pico alguns anos depois (2001-03) mas também diminuiu drasticamente desde entãow5.
References
- Franscini N et al. (2006) Prion protein in milk. PLoS ONE 1: e71. doi: 10.1371/journal.pone.0000071
- All PLos ONE articles are freely available online.
- Gatti JL et al. (2002) Prion protein is secreted in soluble forms in the epididymal fluid and proteolytically processed and transported in seminal plasma. Biology of Reproduction 67: 393-400. doi: 10.1095/biolreprod67.2.393
- Ligios C et al. (2005) PrPSc in mammary glands of sheep affected by scrapie and mastitis Nature Medicine 11: 1137–1138. doi: 10.1038/nm1105-1137
- Mortimer D, Barratt CLR (2006) Is there a real risk of transmitting variant Creutzfeldt–Jakob disease by donor sperm insemination? Reproductive BioMedicine Online 13: 778–790
- To view articles in Reproductive BioMedicine Online, it is necessary to register, but registration is free. See: http://www.rbmojournal.com/
- Si K et al. (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 140: 421-435. doi: 10.1016/j.cell.2010.01.008
- Wang F et al. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327: 1132-1135. doi: 10.1126/science.1183748
Web References
- w1 – Informações sobre o kuru estão disponíveis no website do ‘US National Institute of Neurological Disorders and Stroke’: www.ninds.nih.gov/disorders/kuru
- Em 1976, Carleton Gajdusek recebeu o Prémio Nobel da Medicina pelo seu trabalho sobre o kuru. Mais informações estão disponíveis no comunicado de imprensa, na sua autobiografia, no discurso de Prémio Nobel e outros materiais no website do Prémio Nobel. Ver: http://nobelprize.org/nobel_prizes/medicine/laureates/1976
- w2 – Para saber mais sobre o trabalho de Stanley Prusiner’s, ver o comunicado de imprensa, a sua autobiografia e outros materiais no website dos Prémios Nobel: http://nobelprize.org/nobel_prizes/medicine/laureates/1997
- w3 – Estatísticas dos casos registados de BSE e mortes provocadas por vCJD estão disponíveis no website do ‘European Centre for Disease Prevention and Control’: (www.ecdc.europa.eu) ou através do link directo: http://tinyurl.com/yjbx8tn
- w4 – Para saber mais sobre a predisposição genética para adquirir doenças priónicas, ver o website da unidade de priões do ‘Medical Research Council’ do Reino Unido (www.prion.ucl.ac.uk) ou use o link directo: http://tinyurl.com/yaqau4a
- w5 – Estatísticas sobre BSE e muitas outras doenças animais estão disponíveis no website da ‘World Organisation for Animal Health’: www.oie.int
Resources
- Mais informação sobre priões está disponível nos websites dos ‘US Centers for Disease Control and Prevention’: www.cdc.gov/ncidod/dvrd/prions
- Para saber mais sobre BSE, ver o website da ‘World Health Organization’: www.who.int/zoonoses/diseases/bse/en
- Trabalhos de pesquisa que podem ter interesse incluem:
- Aguzzi A, O’Connor T (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery 9: 237-248. doi: 10.1038/nrd3050
- Collinge, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318: 930-936. doi: 10.1126/science.1138718
- Di Guardo G, Marruchella G (2010) Prions and neuronal death. Cell Death and Disease 1: e6. doi: 10.1038/cddis.2009.9
- Welberg L (2010) Prions: a protective role for prions. Nature Reviews Neuroscience 11: 151. doi: 10.1038/nrn2812
- Os postulados de Koch foram publicados nos finais do século XIX por Robert Koch. Para uma análise mais actual dos postulados e da sua validade, ver:
- Evans AS (1976) Causation and disease: the Henle-Koch postulates revisited. The Yale Journal of Biology and Medicine 49: 175-195. The article can be downloaded free of charge from PubMed Central (www.ncbi.nlm.nih.gov/pmc) or via the direct link: http://tinyurl.com/y9t2pd8
Review
Muitas pessoas ouviram falar da vCJD, ou doença das vacas loucas, mas pouco sabem sobre o agente causador e da doença propriamente dita. Este artigo explica a doença e cita investigação actual sobre o agente que a causa, um prião. Será útil para o tópico de doenças infecciosas incluído em muitos curricula de ciência e para cultura geral noutros assuntos. Os links para dados sobre BSE poderiam ser usados em aulas de matemática ou para aprender a construir gráficos a partir dos dados. O artigo presta-se a debates sobre segurança alimentar, o efeito do surto da doença entre os produtores de carne britânicos, assim como trabalhos de compreensão e aplicação. Algumas ideias para questões de compreensão incluem:
- Há quantos anos estão os priões a ser investigados?
- 10 anos
- 25 anos
- 30 anos
- 40 anos
- Quem ganhou o Prémio Nobel pela investigação realizada com priões?
- O que é um prião?
- Descreva os efeitos das encefalopatias espongiformes.
- Em que tecidos foram descobertos os priões?
- Como conseguiram os priões propagar-se?
Ideias para questões de aplicação são:
- Investigue o surto de BSE no Reino Unido nos finais de década de 80 do séc. XX e descreva as medidas que foram introduzidas para o limitar.
- Formule os postulados de Koch. Explique por que os priões não encaixavam nesta hipótese até recentemente.
Shelley Goodman, Reino Unido