Supporting materials
Bioinformática com papel e lápis: construção de uma árvore filogenética (Word)
Download
Download this article as a PDF





Traduzido por: Artur Melo. A bioinformática é normalmente estudada com computadores potentes. Com a ajuda de Cleopatra Kozlowski, no entanto, pode investigar a nossa ascendência primata – armado apenas com um lápis e um papel.
Como resultado de avanços tecnológicos recentes, é relativamente rápido e fácil determinar uma sequência de ADN ou de uma proteína. Estas sequências por si só, como é óbvio, não são significativas: GAATCCA, por exemplo. Precisamos saber o que essas sequências significam. Que proteínas são codificadas por essa sequência; codificará a sequência realmente uma proteína? Que influência tem uma pequena alteração na sequência de ADN, na estrutura da proteína codificada? Que função desempenha essa proteína na célula? E, é claro, o que pode a sequência de ADN dizer-nos acerca da nossa história evolutiva?
Estas e outras importantes questões biológicas podem ser abordadas pela bioinformática: essencialmente, pela comparação de sequências de ADN e de proteínas – por exemplo, comparando sequências recentemente descobertas com sequências para as quais já temos bastante informação (será que têm uma função semelhante?) ou comparando sequências semelhantes em diferentes espécies.
A bioinformática é, evidentemente, realizada habitualmente com o auxílio de um computador potente. No entanto, é bastante fácil deixar que o computador realize todo o trabalho sem perceber os príncipios subjacentes envolvidos. Por isso, estas actividades estão planeadas para ser feitas em papel, para permitir aos alunos perceber como a análise bioinformática funciona.
Este artigo inclui uma actividade de um grupo de quatro. As duas actividades introdutórias (‘Gene finding’ e ‘Mutations’) e a actividade de conclusão (‘Mobile DNA’) podem ser descarregadas no website do ‘European Learning Laboratory for the Life Sciences (ELLS)w1. Todos os quadros necessários para os alunos realizarem esta actividade, assim como o procedimento passo-a-passo e as respostas às questões, podem ser descarregadas no website da Science in Schoolw2.
A acumulação de mutações provoca a alteração das sequências de ADN ao longo das gerações. A actividade que se segue demonstra como isso pode ser usado para deduzir relações evolutivas entre organismos. Demora cerca de 90 minutos e apenas é preciso um lápis e os quadros, que podem ser obtidos no website da Science in Schoolw2.
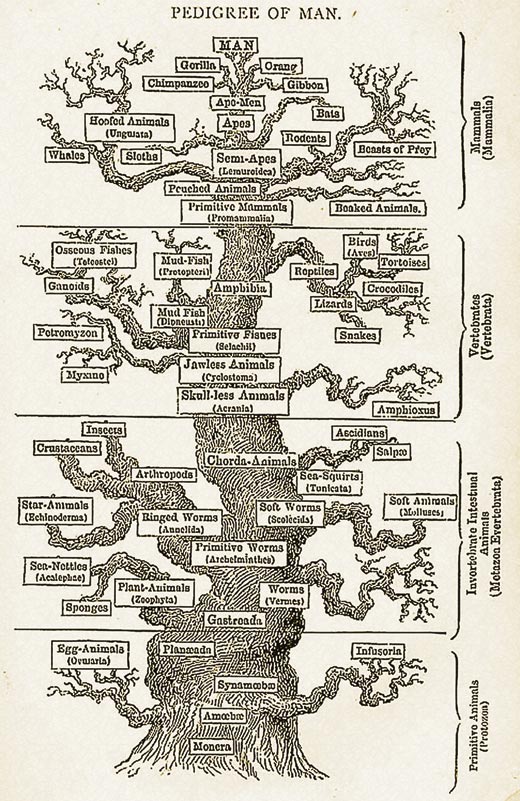
Pense como iria classificar vários animais. Tradicionalmente são usadas diferenças físicas entre os organismos para deduzir relações evolutivas entre eles, por exemplo, se um organismo tem coluna vertebral ou se tem asas. No entanto, este método pode originar problemas. Por exemplo, aves, morcegos e insectos têm asas, mas estarão intimamente relacionados? Como podemos medir há quanto tempo os organismos divergiram a partir de um ancestral comum?
Sabemos, a partir de estudos de sequências de ADN, que as mutações no ADN ocorrem ao acaso a uma taxa muito baixa e são transmitidas dos pais para os filhos. Assim, se assumirmos que todos os organismos têm um ancestral comum, podemos usar as diferenças nas sequências homólogas para medir quanto tempo decorreu desde que os organismos divergiram. Por outras palavras, quanto mais tempo passou desde que duas espécies divergiram a partir de um ancestral comum, mais diferenças haverá nas suas sequências de ADN.
Define-se sequências homólogas como sequências que em dois organismos têm uma origem comum. Na realidade, não há provas seguras que quaisquer duas sequências são homólogas (não estávamos lá para ver o ADN a mudar ao longo do tempo) mas se forem suficientemente semelhantes, assume-se normalmente que são ‘homólogas’. Para conhecer quão semelhantes são duas sequências, precisamos de as alinhar correctamente (isto não faz parte desta actividade).
Tenha em atenção que regiões diferentes de ADN – regiões codificantes e não-codificantes – evoluem a taxas diferentes. Geralmente, as regiões codificantes evoluem mais lentamente, porque uma mutação que provoca alterações numa proteína é normalmente mais agressiva para o organismo – é menos provável que sobreviva e produza descendência. Este aspecto é discutido na actividade ‘Mobile DNA’..
Para ilustrar o conceito de homologia, podemos usar o exemplo da filologia – o estudo da evolução das línguas. De facto, há muitas semelhanças entre os métodos usados no estudo da evolução da língua e dos organismos.
Usar as diferenças entre sequências de fragmentos de ADN é um pouco como comparar uma palavra com o mesmo significado em diferentes línguas, para verificar a proximidade da sua relação.
| Arménio | gatz |
| Basco | katu |
| Holandês | kat |
| Inglês | cat |
| Estónio | kass |
| Finlandês | kissa |
| Islandês | kottur |
| Italiano | gatto |
| Norueguês | katt |
| Polaco | kot |
| Português | gato |
| Russo | kot |
| Espanhol | gato |
| Sueco | katt |
Podemos ver que as palavras para ‘gato’ em Italiano, Espanhol e Português são quase iguais: gatto, gato e gato. Tanto em Sueco como em Norueguês a palavra é ‘katt’ mas em Finlandês é diferente: ‘kissa’. Apesar de, tal como a Suécia e a Noruega, a Finlândia ser um país nórdico, a palavra finlandesa para ‘gato’ é mais parecida com a palavra da Estónia, ‘kass’. De facto, as duas línguas estão intimamente relacionadas. Assim podemos aprender um pouco sobre relações entre línguas estudando como as palavras se foram modificando ao longo do tempo.

Nesta actividade, vamos construir uma árvore filogenética usando cinco sequências de ADN homólogas de primatas. Como as sequências foram preparadas, não podemos deduzir nenhuma estimativa da distância genética; para criar uma árvore filogenética significativa a partir de dados reais precisávamos de sequências bastante mais longas. Não obstante, as sequências fictícias (no Quadro 2) foram seleccionadas para fornecer uma perspectiva precisa das relações entre os primatas.
Nota: todos os quadros necessários para os alunos completarem esta actividade podem ser obtidos no website da Science in Schoolw2.
| Primata | Sequence |
|---|---|
| Neandertal (n) | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
| Humano (h) | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Chimpanzé (c) | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
| Gorila (g) | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
| Orangotango (o) | ACAACCTGCACTCCTATTCTGCCGAGCCGGGCGCGTGGCAAAGTCC |
| Neandertal | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
|---|---|
| Humano | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Chimpanzé | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
|---|---|
| Gorila | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
Quadros comparativos para todos os pares de espécies, e o quadro preenchido com as diferenças nas sequências (Quadro 4), podem ser obtidos no website da Science in Schoolw2.
| Neandertal | Humano | Chimpanzé | Gorila | Orangotango | |
|---|---|---|---|---|---|
| Neandertal | 0 | 3 | |||
| Humano | 3 | 0 | |||
| Chimpanzé | 0 | 11 | |||
| Gorila | 11 | 0 | |||
| Orangotango |
O número de diferenças de nucleótidos entre duas sequências, dividido pelo número total de nucleótidos em cada sequência (neste caso, 46) dá-nos a distância proporcional entre as duas sequências.
Assume-se que a ‘sequência média’ de duas espécies corresponde ao seu ancestral. Neste exercício, não calculamos directamente a sequência média de, por exemplo, neandertais e humanos, mas a distância evolutiva entre o ancestral de neandertais/humanos e todos os outros primatas do grupo.
| Neandertal/ humano | Chimpanzé | Gorila | Orangotango | |
|---|---|---|---|---|
| Neandertal/ humano | 0 | (4+5)/2 = 4.5 | (11+12)/2=11.5 | |
| Chimpanzé | (4+5)/2 = 4.5 | 0 | ||
| Gorila | (11+12)/2=11.5 | 0 | ||
| Orangotango |
Usando o Quadro 5, pode começar a construir a árvore evolutiva.
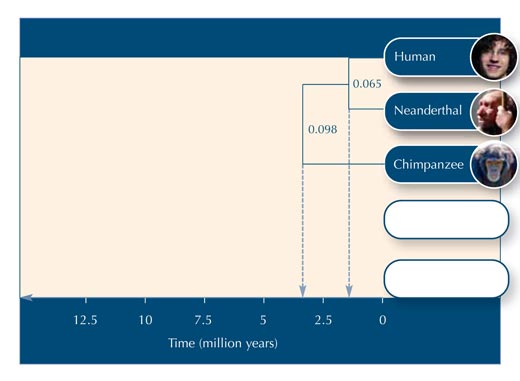
Consideremos que são necessários 20 milhões de anos para que cada um dos nucleótidos desta sequência de ADN se altere. Assim, para que a sequência de ADN se altere 0.065, seriam necessários 0.065*20 milhões = 1.3 milhões de anos. O ramo deve, por isso, medir 1.3 milhões de anos na escala do tempo(ver Figura 2).
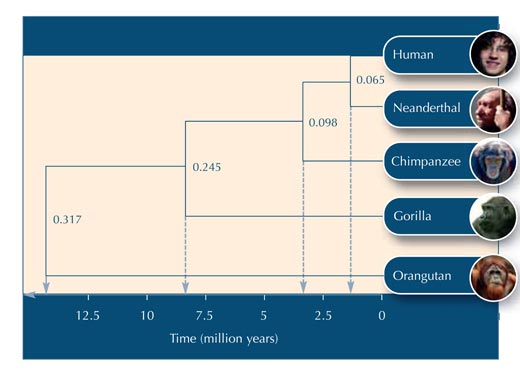
Seguem-se algumas questões que pode usar para avaliar se os seus alunos compreenderam esta actividade. As respostas podem ser obtidas no website da Science in Schoolw2.
Esta actividade foi desenvolvida numa colaboração especial entre o ‘European Learning laboratory for the Life Sciences (ELLS)w1 e o ‘European Molecular Biology Laboratory’s E-STAR Fellows’ para criar recursos educativos para as escolas. Cleopatra Kozlowski foi apoiada pela E-STAR fundada pela ‘European Commission’s Framework Programme 6 Marie Curie Host Fellowship for Early Stage Research Training’, com o contrato número MEST-CT-2004-504640.
Hodge R (2006) A new tree of life. Science in School 2: 17-19.
Quando pensamos na bioinformática, provavelmente imaginamos enormes computadores e equipamento de sequenciação, mas os métodos desta ciência recente podem ser apresentados como actividades didáticas simples, a realizar com papel e lápis, como Cleopatra Kozlowski faz neste artigo.
A autora desafia-nos a construir a árvore filogenética de humanos e outros primatas tendo como base as diferenças genéticas entre pequenas sequências de ADN (fictícias). A actividade proposta pode ser proveitosamente (e agradavelmente) explorada em escolas secundárias para abordar alguns tópicos de biologia tais como a utilização de relógios moleculares no estudo da evolução.
O artigo está orientado para professores de ciências, incluindo exercícios de avaliação no final do texto; os alunos podem usar as questões para aprofundar o conhecimento sobre o assunto. As referências web fornecem mais informações e recursos.
Giulia Realdon, Itália
Bioinformática com papel e lápis: construção de uma árvore filogenética (Word)
Download this article as a PDF