Biologinių duomenų bazių naudojimas mokant evoliucijos ir biochemijos Teach article
Vertė Urtė Neniškytė. Internetiniai įrankiai gali būti naudojami palyginti baltymų sekoms ir suprasti, kaip išsivystė įvairūs organizmai.
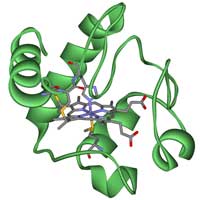
struktūra.
Paveikslas publikuojamas
Klaus Hoffmeier/Wikimedia
commons leidimu
Praeityje mokslininkai evoliucinius tyrimus atlikdavo lygindami fizines rūšių charakteristikas – žinomas kaip jų fenotipai – aptiktas suakmenėjusiose iškasenose. Tačiau atradus molekulinį laikrodį, visa tai pasikeitė.
Molekulinio laikrodžio idėja kilo pastebėjus, kad kuo ilgesnis laikas skiria dvi rūšis, atsiskyrusias nuo bendro protėvio, tuo labiau skiriasi jų DNR ar baltymų sekos (apžvelgta Bromham ir Penny, 2003).
Lyginant homologinius – kitaip tariant, priklausančius dviem organizmams, turintiems bendrą protėvį – genus ar baltymus, galima išmatuoti, kiek laiko praėjo nuo tada, kai organizmai atsiskyrė. Tai galima pavaizduoti filogenetiniu medžiu.
Norint palyginti, kiek panašūs du genai, reikia turėti jų sekas ir jas tinkamai sugretinti (Kozlowski, 2010). Anksčiau gauti šias sekas buvo labai sudėtinga, tačiau dabar taip nebėra.
Jūsų mokiniai tikriausiai jums sakė, kad viską galima rasti internete – šįkart jie teisūs. Internete daug laisvai prieinamų biologinių duomenų bazių su tikrais mokslinių tyrimų duomenimis, tačiau šioje užduotyje naudosime du išskirtinius šaltinius.
Nacionalinis biotechnologinės informacijos centras (National Center for Biotechnology Information, NCBI)w1 iš Bethesdos, MD, JAV suteikia prieigą prie biomedicininės ir genominės informacijos, o Europos bioinformatikos institutas (European Bioinformatics Institute, EBI)w2, esantis Hinxtone, JK, suteikia laisvai prieinamus gyvybės mokslų eksperimentinius duomenis ir atlieka pagrindinius skaičiuojamosios biologijos tyrimus. NCBI duomenų bazėje rasite bet kurią jau nustatytą geno ar baltymo seką, o tuomet, naudodami EBI įrankius, galėsite sekas palyginti ir ištirti.
Mokomoji užduotis
Tyrinėjant evoliucinius ryšius tarp skirtingų organizmų, svarbu atidžiai pasirinkti naudojamus genus ar baltymus. Yra žinoma keletas tinkamų homologinių genų, koduojančių tokius baltymus kaip hemoglobinas ar citochromas c, ir šioje užduotyje mes naudosime pastarąjį. Citochromas c yra mažas hemo grupę turintis baltymas, kuris yra centrinis elektronų pernašos grandinės komponentas mitochodrijose. Visi aerobiniai organizmai išsivystė iš bendro protėvio, kuris pirmasis panaudojo citochromą c, todėl šis baltymas yra tinkamas mūsų tikslamsw3.
Ši mokomoji užduotis sudaryta iš trijų skirtingų dalių:
- surasti skirtingų organizmų citochromų c aminorūgščių sekas,
- jas sugretinti ir
- sudaryti filogenetinį medį.
Galiausiai pateikiama keletas klausimų, kurie padės tyrinėjant evoliucinius ryšius.

spragtelėkite ant paveikslo
Baltymų sekų paieška
- Atsiverskite NCBI tinklalapįw1.
- Paieškos laukelyje puslapio viršuje iš išskleidžiamojo sąrašo pasirinkite protein (liet.baltymas).
- Įrašykite rūšies pavadinimą, pvz., Homo sapiens, ir cytochrome c (liet. citochromas c).
- Spragtelėkite paieškos mygtuką.
- Naujame puslapyje bus pateikti jūsų paieškos rezultatai. Dauguma jų yra ta pati seka iš skirtingų šaltinių, tačiau kai kurie gali būti dalinės sekos arba priklausyti kitoms rūšims ar baltymams. Atidžiai pasirinkite tinkamą dominančio baltymo seką ir spragtelėkite ant apačioje esančios nuorodos FASTA.
- Atsidariusiame puslapyje nukopijuokite didžiųjų raidžių eilutę, žyminčią aminorūgščių seką. Įterpkite raides į Word dokumentą, nepamirškite pasižymėti, kuriam organizmui ši seka priklauso.
- Pakartokite tą patį keletui skirtingų organizmų, priklausomai nuo to, ką norite tyrinėti su savo mokiniais. Galite įtraukti skirtingus primatus, kad išsiaiškintumėte, kaip išsivystė žmogus, arba organizmus iš penkių tradicinių karalysčių, kad sužinotumėte, kaip vystėsi gyvybė apskritai. Šioje priemonėje naudojami 3 gyvūnai, 2 augalai, 2 dumbliai, grybas ir pirmuonis.
Sekų sugretinimas
- Atsiverskite Europos bioinformatikos instituto (EBI) svetainęw2 ir spragtelėkite ant Services(liet. paslaugos). Tuomet pasirinkite proteins(liet. baltymai).
- Spragtelėkite ant Clustal Omega. Nukopijuokite tekstą iš savo Word dokumento ir įterpkite jį į teksto laukelį, pažymėtą STEP 1 (liet. 1 ŽINGSNIS).
- STEP 2 (liet. 2 ŽINGSNIO) laukelyje pasirinkite sugretinimo rezultatų pateikimo formatą, tokį kaip Clustal w/ numbers (liet.Clustal su skaičiais), kuris pateiks kiekvienos sekos ilgį. Galiausiai spragtelėkite ant Submit(liet. pateikti), kad užbaigtumėte STEP 3 (liet. 3 ŽINGSNĮ).
- Keleto sekų sugretinimas bus pateiktas naujame lange. Pirmiausia, galite spragtelėti, kad būtų rodomos spalvos. Ši parinktis kiekvienai aminorūgščiai paskirs po spalvą, kad jas būtų lengviau atpažinti.
- Tyrinėdami sugretinimą, turėkite omenyje šiuos ženklus: žvaigždutė (*) reiškia, kad sekos toje padėtyje yra identiškos; dvitaškis (:) nurodo konservatyvius pakeitimus (ta pati spalvų grupė); taškas (.) žymi pusiau konservatyvius pakeitimus (panašios formos). Spalvos sugrupuoja aminorūgštis pagal jų savybes. Raudonos yra mažos, hidrofobinės, aromatinės; mėlynos yra rūgštinės; avietinės yra bazinės; žalios yra bazinės, turinčios hidroksilo, amino, amido grupes; pilkos yra visos kitos.
- Jeigu spragtelėsite ant parinkties Result Summary (liet. Rezultatų santrauka), galėsite peržiūrėti, kokia procentinė identiškumo dalis, išsaugota skirtinguose organizmuose, buvo nustatyta sugretinus. Šioje matricoje galite rasti, koks yra procentinis dviejų organizmų identiškumas pagal citochromo c seką. Be to, jeigu jūsų kompiuteryje įdiegta Java™ programa, galite naudoti Jalview – nemokamą programą, skirtą redaguoti, vaizdinti ir analizuoti kelių sekų sugretinimą. Su Jalview galėsite peržiūrėti tapačias citochromo c sekų dalis ir kiek konservatyvios yra skirtingos aminorūgštys.
Clustal Omega programinė įranga turi daug skirtingų parinkčių, kuriose taikomi sudėtingesni matematikos principai, nei reikia mūsų tikslams. Jei norite apie Clustal Omega sužinoti daugiau, žr. Sievers et al. (2011) straipsnį.
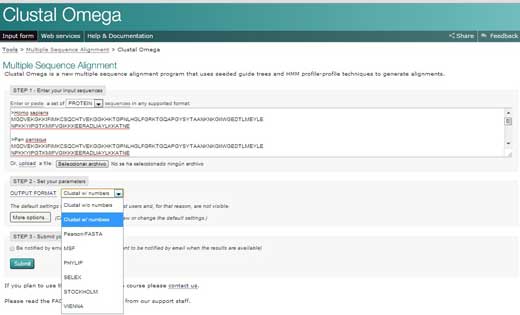
spragtelėkite ant paveikslo
Filogenetinio medžio sudarymas
- Clustal Omega rezultatų apačioje spragtelėkite ant Phylogenetic tree (liet.filogenetinis medis) (jums reikės turėti įdiegtą Java™ programą).
- Galite sudaryti filogenetinį arba kladogramos medį. Kladogramoje medžio šakų ilgiai yra pasirinktiniai, o filogenetinio medžio šakų ilgiai nurodo, kiek bėgant laikui pakito baltymas.
Aptarti papildomai
spragtelėkite ant paveikslo
- Homologinės molekulės yra divergentinės evoliucijos pavyzdys. Kaip galėtumėte paaiškinti divergentinę evoliuciją naudodami citochromą c?
- Sugretinimus galima atlikti naudojant nukleotidų (genų) ar aminorūgščių (baltymų) sekas. Kaip manote, kodėl tyrinėjant evoliucinius ryšius naudingiau naudoti baltymus, o ne DNR?
- Filogenetiniuose medžiuose „kladą“ sudaro visi organizmai, kurie turi bendrą protėvį. Pateikite pavyzdį iš savo kladogramos.
- Kurių organizmų rūšių vėliausią atsiskyrimą rodo citochromo c filogenetinė analizė? Koks yra bendras rūšių atsiskyrimų skaičius?
- Kaip manote, kodėl mutacijos pakeitė vienas aminorūgštis, o ne kitas? Ar manote, kad konservatyvios aminorūgštys visai nepakito todėl, kad jų kodonuose neįvyko jokių mutacijų?
- Nurodykite keletą tokių konservatyvių aminorūgščių savo sugretinime. Naudodamiesi internetu, ištirkite jų funkciją.

Žodynėlis
Kladograma – šakota diagrama, kurioje vaizduojami evoliuciniai ryšiai tarp rūšių; šakų ilgiai pasirinktiniai.
Konsensuso seka – žinomas konservatyvių sekų rinkinys arba apskaičiuota dažniausių aminorūgščių tvarka, nustatoma kiekvienoje sugretintų sekų padėtyje.
Konservatyvi aminorūgščių seka – polipeptido aminorūgščių seka, kuri yra panaši skirtinguose organizmuose.
FASTA – tekstinis formatas, skirtas pateikti nukleotidų ar peptidų sekoms, naudojant vienos raidės kodus. FASTA formato seka prasideda vienos eilutės aprašymu, po kurio eina sekos duomenų eilutės. Aprašymo eilutė išskirta iš sekos duomenų ženklu daugiau (>).
Homologiniai baltymai – baltymai, nustatomi kai kuriuose organizmuose, kilusiuose iš to paties protėvio.
Filogenetinis medis – šakota diagrama, kurioje vaizduojami evoliuciniai ryšiai tarp rūšių; šakų ilgiai nurodo skirtumą tarp dviejų baltymų ar genų.
Rūšių susiformavimas – akimirka, kai nuo tėvinės rūšies atsiskiria naujos rūšys.
Padėka
Autorius dėkoja savo kolegei María Isern, padėjusiai suredaguoti angliško straipsnio kalbą.
References
- Bromhan L. Penny D. (2003) The modern molecular clock. Nature Reviews Genetics 5: 216-224
- Kozlowski C. (2010) Bioinformatics with pen and paper: building a phylogenetic tree. Science in School 17: 28-33.
- Sievers F. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7: 539
- Šis straipsnis laisvai prieinamas Molecular Systems Biology svetainėje.
Web References
- w1 – JAV Nacionalinis biotechnologinės informacijos centras (US National Center for Biotechnology Information) suteikia prieigą prie biomedicininės ir genominės informacijos.
- w2 – Europos bioinformatikos institutas (European Bioinformatics Institute) suteikia laisvai prieinamus gyvybės mokslų eksperimentinius duomenis, atlieka pagrindinius skaičiuojamosios biologijos tyrimus ir remia akademinius bei pramonės tyrėjus, siūlydami plačią naudotojų ugdymo programą.
- w3 – John Kimball parašė internetinį vadovėlį Taxonomy: Classifying Life (liet.Taksonomija: gyvybės klasifikavimas), kuriame yra skyrius Phylogenetic trees (liet. Filogenetiniai medžiai).
Resources
- Kalifornijos universiteto Paleontologijos muziejaus svetainėje Understanding Evolution (liet. Suprask evoliuciją) pateikiama labai geros informacijos, kaip sudaromi ir vertinami filogenetiniai medžiai.
- Daugiau informacijos apie citochromo c naudojimą filogenetiniuose medžiuose.
- Protein Data Bank (liet. Baltymų duomenų banko) svetainė Europoje priklauso Europos bioinformatikos institutui, jame galima surasti ir peržiūrėti trimatę citochromo c struktūrą.
- Tree of Life (liet. Gyvybės medžio) svetainėje galite interaktyviai tyrinėti ir peržiūrėti sero Davido Attenborough vedamą televizijos programą Tree of Life, kuri buvo rodoma BBC kanalu.
Review
Biologijos mokytojai galėtų naudoti šį straipsnį siekdami susieti evoliucinės biologijos, mokslo istorijos, biochemijos ir genetikos temas. Siekiant šį straipsnį panaudoti kiek galima naudingiau, svarbu, kad mokiniai suprastų DNR ir baltymų biochemijos pagrindus.
Straipsnyje aprašyta užduotis svarbi motyvuojant mokinius savarankiškai atlikti tikrus tyrimus naudojant mokslines duomenų bazes. Mokomojoje klasėje mokiniai, dirbdami nedidelėmis grupėmis, turėtų palyginti baltymų, tokių kaip citochromas c, ar DNR sekas ir išsiaiškinti skirtumus tarp filogenetinių ir kladogramų medžių. Bioinformatika yra labai naudinga vidurinėje mokykloje kaip įvairių lygių „integruoto turinio ir kalbos lavinimo“ įrankis, kurį kalbų, istorijos ir fizikos mokytojai gali pritaikyti tarpdalykiniuose projektuose. Šis straipsnis, siejantis šiuos skirtingus dalykus, taip pat galėtų būti naudojamas pradėti diskusijai apie pažangos ir tokio tipo mokslinių tyrimų ribas.
Marina Minoli, didaktikos ekspertė, Agora universiteto centras (Agora University Centre), Italija