Supporting materials
Bioinformatika tollal és papírral: készítsünk filogenetikai fát (Word)
Download
Download this article as a PDF





Fordította: Kapitány János Sándor. A bioinformatikához általában egy erős számítógépre van szükségünk. Azonban Cleopatra Kozlowski segítségével csupán egy tollal és papírral felfegyverkezve is megvizsgálhatjuk főemlős származásunkat.
A legújabb technológiáknak köszönhetően, viszonylag könnyen és gyorsan meghatározhatjuk egy DNS vagy fehérje szekvenciáját. Ezek a szekvenciák önmagukban természetesen elég keveset árulnak el számunkra: például GAATCCA. Tudnunk kell értelmezni ezeket a szekvenciákat. Melyik fehérjét kódolja ez a DNS szekvencia; egyáltalán fehérjét kódol-e az adott szekvencia? Milyen hatással van a DNS szekvencia kis változtatása a kódolt fehérje szerkezetére? Milyen funkciója less a fehérjének a sejtben? És, persze mit tud mesélni számunkra a DNS szekvenciánk az evolúciós történetünkről?
Ezekkel és más fontos biológiai kérdésekkel foglalkozik a bioinformatika: különösen, a DNS vagy fehérjék szekvenciájának összehasonlításával – például újonnan felfedezett szekvenciák összehasonlítása olyan, korábban már megismert szekvenciákkal, amelyekről már több információval is rendelkezünk (talán van hasonló funkciójuk?) vagy összehasonlíthatjuk a különböző fajokban található hasonló szekvenciákat.
A bioinformatika, normális esetben egy erős számítógép segítségével működik. Az azonban túlságosan könnyű lenne, ha hagynánk, hogy egy számítógép végezze el az összes munkát anélkül, hogy megértenénk a lefektetett alapelveket. Ebből kifolyólag ezt a tevékenységet papíron végezzük el, hogy a diákok megérthessék, hogyan működik a bioinformatikai analízis.
Ez a cikk egy négyféle tevékenységet tartalmazó csoport egyike. A két bevezető tevékenység (‘Gén keresés’ és ‘Mutációk’) és a befejező tevékenység (‘Mobil DNS’) letölthető az Az Európai Tanulási Laboratórium a Természettudományok számára (European Learning Laboratory for the Life Sciences = ELLS)w1. A tanulók számára szükséges táblázatok, együtt az eljárás lépésről-lépésre leírásával és a megértést segítő kérdésekre adott válaszokkal, letölthetők a Science in School weboldalárólw2.
A mutációk felhalmozódása okozza a DNS szekvencia generációk alatt bekövetkező változását. A következő tevékenység azt mutatja meg, hogyan lehet ezt a szervezetek közötti evolúciós kapcsolatok levezetésére használni. Körülbelül 90 percet vesz igénybe és nem igényel semmi mást csak egy tollat és a táblázatokat, amelyek letölthetők a Science in School weboldalárólw2.
Gondoljatok bele, hogyan kellene osztályozni a különböző állatokat. Hagyományosan, a szervezetek közötti fizikai különbségeket (morfológiai jegyeket) használták az evolúciós kapcsolataik levezetése céljából, például hogy egy szervezetnek van hátgerince vagy vannak szárnyai. Ez problémákat okozhat. Például a madarak, denevérek és rovarok mindegyike rendelkezik szárnyakkal, de valóban közeli rokonok? Hogyan mérnéd, hogy a jelenlegi szervezetek, hogyan tértek el a korábbi közös őstől?
A DNS szekvenálási tanulmányainkból tudjuk, hogy a DNS mutációk véletlenszerűen fordulnak elő, nagyon kis arányban átadódnak a szülőktől az utódnak. Így ha feltételezzük, hogy minden szervezetnek van egy közös őse, akkor arra használhatjuk a homológ szekvenciák különbségeit, hogy megmérjük, milyen hosszúságúak lettek, amióta a szervezetek eltértek egymástól. Más szavakkal, ha két faj régebben jött létre egy közös ősből, akkor a DNS szekvenciájuk jobban eltér egymástól.
A homológ szekvenciákat úgy határozhatjuk meg, mint két azonos eredetű szervezetben található szekvenciák. Valójában nincs bizonyítékunk arra, hogy bármely két szekvencia homológ lenne (mivel nem voltunk ott, hogy megfigyeljük a DNS-t, ahogy az idő során megváltozott) de amelyek kellően hasonlóak, azokról gyakran feltételezik, hogy homológok. Két szekvencia hasonlóságát akkor tudjuk megvizsgálni, ha azokat helyesen rendezzük el egymáshoz képest (de ez nem része ennek a tevékenységnek).
A különböző DNS régiók – kódoló (exon) és nem-kódoló (intron) szakaszok – eltérő sebességgel fejlődnek. Általában a kódoló régiók sokkal lassabban változnak, mert egy mutáció, ami a fehérjében változást okoz, általában sokkal inkább a szervezetben vált ki hatást – kevésbé él túl marad fenn az utódban. Erről a ‘Mobil DNS’ tevékenységben lehet beszélgetni.
A homológia fogalmának illusztrálásához, felhasználhatjuk példának a filológiát – a nyelvek evolúciójának tanulmányozását. Tény, hogy sok párhuzamot találhatunk a nyelvek és a szervezetek evolúciójának tanulmányázásában.
A DNS szekvenciák töredékei közötti különbségek értelmezése, egy kicsit olyan mintha összehasonlítanánk egy adott szónak különböző nyelvekben előforduló alakjait, hogy lássuk milyen közeli a kapcsolatuk egymáshoz.
| Örmény | gatz |
| Baszk | katu |
| Holland | kat |
| Angol | cat |
| Észt | kass |
| Finn | kissa |
| Izlandi | kottur |
| Olasz | gatto |
| Norvég | katt |
| Lengyel | kot |
| Portugál | gato |
| Orosz | kot |
| Spanyol | gato |
| Svéd | katt |
Láthatjátok, hogy a ‘macska’ szó az olaszban, spanyolban és portugálban majdnem ugyanaz: gatto, gato és gato. Mind a Svéd és Norvég nyelvben ‘katt’, de a Finn nyelvben eltérő: ‘kissa’. Habár Svédország, Norvégia és Finnország mind Északi ország, a Finn szó a ‘macska’ esetében inkább az Észt ‘kass’ szóhoz hasonlít. Tény, hogy ez a két nyelv sokkal szorosabb kapcsolatban van egymással. Szóval lehet egy kicsit tanulnia nyelvek kapcsolatának tanulmányozásából arról, hogyan változtak a szavak az idők során.

Ezen tevékenység során, egy filogenetikai fát fogunk készíteni öt főemlős homológ DNS szekvenciájának segítségével. Mivel ezek a szekvenciák kitaláltak, ezért nem tudunk következtetni valódi becsült genetikai távolságokra; egy értelmes filogenetikai fa létrehozásához valós adatokból, és sokkal hosszabb szekvenciákból kellene építkeznünk. Mindazonáltal ezeket a fiktív szekvenciákat (a 2. táblázatban) úgy választottuk meg, hogy ésszerűen pontos képet adjanak a főemlősök kapcsolatáról.
Megjegyzés: A tanulók számára a tevékenység teljesítéséhez szükséges táblázatok letölthetők a Science in School weboldalárólw2.
| Főemlős | Szekvencia |
|---|---|
| Neander-völgyi (n) | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
| Emberi (h) | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Csimpánz (c) | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
| Gorilla (g) | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
| Orángután (o) | ACAACCTGCACTCCTATTCTGCCGAGCCGGGCGCGTGGCAAAGTCC |
| Neander-völgyi | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
|---|---|
| Emberi | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Csimpánz | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
|---|---|
| Gorilla | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
Az összehasonlító táblázatok minden szekvencia párhoz és a főemlősök szekvenciáinak különbségeit tartalmazó befejező táblázat (4. táblázat) letölthető a Science in School weboldalárólw2.
| Neander-völgyi | Emberi | Csimpánz | Gorilla | Orángután | |
|---|---|---|---|---|---|
| Neander-völgyi | 0 | 3 | |||
| Emberi | 3 | 0 | |||
| Csimpánz | 0 | 11 | |||
| Gorilla | 11 | 0 | |||
| Orángután |
A nukleotid eltérések száma két szekvenciában osztva a nukleotidok számával szekvenciánként (ebben az esetben 46) megadja az arányos távolságot a két szekvencia között.
Az ‘átlagos szekvenciája’ két fajnak feltételezhetően a közös ősüknek köszönhető. Ebben a feladatban, mi közvetlenül nem számoljuk ki az átlagos szekvenciákat, például a Neandervölgyiek és az emberek között, hanem az evolúciós távolságokat számoljuk ki a Neandervölgyi /emberi ősök és a csoportban szereplő főemlősök között.
| Neander-völgyi és emberi | Csimpánz | Gorilla | Orángután | |
|---|---|---|---|---|
| Neander-völgyi és emberi | 0 | (4+5)/2 = 4.5 | (11+12)/2=11.5 | |
| Csimpánz | (4+5)/2 = 4.5 | 0 | ||
| Gorilla | (11+12)/2=11.5 | 0 | ||
| Orángután |
Az 5. táblázat használatával megkezdhetjük az evolúciós fánk építését.
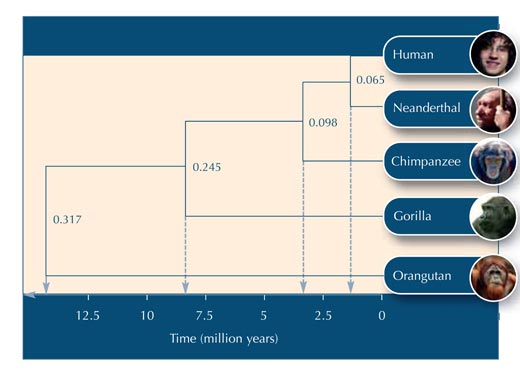
Alább található néhány kérdés, aminek segítségével letesztelhetjük, hogy tanulóink megértették-e a tevékenységet. A válaszok letölthetők a Science in School weboldalárólw2.
Ez a tevékenység az Európai Élettudományi Oktató Laboratórium (European Learning Laboratory for the Life Sciences = ELLS)w1 és az Európai Molekuláris Biológia Laboratórium (European Molecular Biology Laboratory) E-STAR munkatársai közötti különleges kollaborációval készült, tanítási anyagként iskolák számára. Cleopatra Kozlowski egy E-STAR ösztöndíjas volt, ami az Európai Bizottság 6. keretprogramjának Marie Curie ösztöndíja Korai stádiumú kutatási tréningeknek, a MEST-CT-2004-504640 szerződés szám alatt.
Hodge R (2006) A new tree of life. Science in School 2: 17-19.
Amikor bioinformatikára gondolunk általában nagy teljesítményű számítógépekkel és szekvenáló gépekkel képzeljük el, de ennek az új tudománynak az eljárásai bemutathatók egyszerű osztálytermi gyakorlatokkal is, amihez csak ceruza és papír szükséges, ahogy Cleopatra Kozlowski is csinálta ebben a cikkben.
A szerző kihívásként azt tűzi ki számunkra, hogy készítsünk egy olyan filogenetikai fát, amelyben az emberek és más főemlősök közötti távolságok mutatjuk be, amihez genetikai különbségeket használunk, (hamis) rövid DNS szekvenciákat. A javasolt tevékenység hasznosan kiaknázható (és élvezetes) a középiskolákban, néhány trükkös biológia témánál, például az evolúció tanításakor a molekuláris órák témánál.
A cikk célja a tudományos tantárgyakat oktató tanárok segítése, akik a tanítás során használhatják a szövegértési gyakorlatot a szöveg végén; a diákok pedig arra használhatják a kérdést, hogy elmélyítsék ismereteiket a témában. Az idézett internetes hivatkozásokon további információkat és forrásokat találhatunk.
Giulia Realdon, Olaszország
Bioinformatika tollal és papírral: készítsünk filogenetikai fát (Word)
Download this article as a PDF